Project design: qualifying variant evidence standard (QV-ES)
Last update: 20251123
TLDR: Health systems, research, and industry cannot exchange genomic results because each pipeline outputs something different; QV-ES solves this with a shared national-scale rule spec and a minimal evidence layer that every pipeline can generate and every institution can verify.
Why do I need this?

- I am a clinical scientist and I want to confirm quickly if a reported variant is reliable and suitable for accreditation standards.
- I am a data engineer and I want a stable evidence layer that fits cleanly into my data systems.
- I work in industry and I want a shared output that keeps my methods private but still verifiable.
- I am a citizen and I want clear results from cutting edge science and medicine, with my personal data respected.
How is this solved?
System name: QV Evidence Framework
Evidence standard: QV Evidence Standard (QV-ES)
The QV Evidence Standard (QV-ES) is one pillar of a larger framework. The framework consists of three components: the QV rule specification, the QV registry, and the QV-ES, which defines the minimal verifiable evidence required for interpretation in clinical genetics. QV sets give us a clean way to separate genetic analysis variables from the software that uses them.
The rule specification provides the YAML or JSON format, the registry stores both QV rule sets as versioned objects, and QV-ES supplies the evidence rules that pipelines use to produce outputs suitable for relational or graph databases at national scale, such as PostgreSQL and RDF.
Genome analysis providers supply the variant results for interpretation. To verify and trust their result, the framework uses the reverse logic to measuring how much verifiable evidence invalidates an interpretation. Private companies and public research gain a shared evidence metric while keeping internal methods and IP separate.
Technical documentation
The open source design document for this project has been prepared by Switzerland Omics and is available directly at https://docs.switzerlandomics.ch/pages/design_qv_evidence_flag.
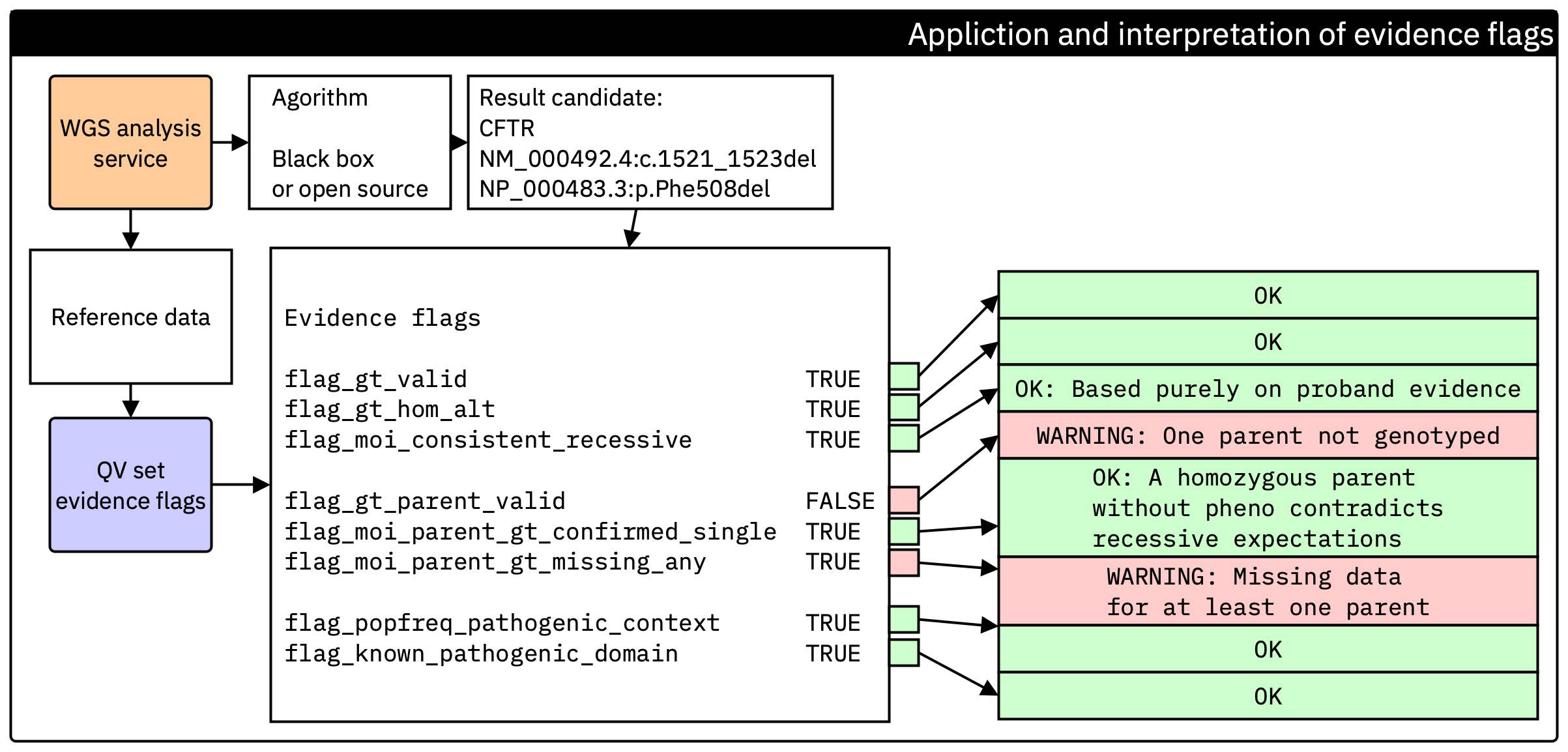 Figure. Generation of the evidence flag set at the end of secondary analysis. Each flag is computed directly from reference data and standard checks, independent of the calling algorithm. These flags are then carried forward into tertiary interpretation so that end users can verify the essential evidence without access to the upstream pipeline.
Figure. Generation of the evidence flag set at the end of secondary analysis. Each flag is computed directly from reference data and standard checks, independent of the calling algorithm. These flags are then carried forward into tertiary interpretation so that end users can verify the essential evidence without access to the upstream pipeline.
Our goal
The SGA is focused on publishing the official guideline manuscript, currently on our release page:
- Guideline: Swiss Genomics Association consensus guideline for evidence-based genomic variant interpretation in Mendelian disease.
About (this page) | PDF (EN) | Repository

